Marguerite Hebert was born around 1652 to Etienne Hebert and Marie Gaudet in or near Port Royal, Acadia.
Marguerite’s life is at least partially documented, but at the same time uncertain, in part because we have conflicting information that has proven impossible to sort through and resolve.

Marguerite Hebert is first found in the 1671 census as the last entry on this page with her recently widowed mother.
Marie Gaudet, widow of Etienne Hebert, 38. She has 10 children, two married children: Marie 20, Marguerite 19; Emmanuel 18, not yet married, Etienne 17, Jean 13, Francoise 10, Catherine 9, Martine 6, Michel 5, Antoine 1, 4 cattle, 5 sheep and 3 arpents of cultivated land
The 1671 census is unique in that married children are listed with their parents, plus in their own households. This is the BEST IDEA EVER, because females are listed by their birth surname, and this census clearly ties them to their parents AND spouse, even without parish records.
Except, that is, for Marguerite.
Of course.
Marie Gaudet’s entry on the census page clearly states that two of her children are married and lists them. Marie Hebert, Marguerite’s older sister by a year is shown in the census entry above Marie Gaudet’s entry, married to Michel de Forest, living in her own household.
Marguerite Hebert’s husband, whoever he might have been, is no place to be found, and neither is Marguerite found in another household.
What gives?
Great question. I wish I knew the answer.
Marguerite’s mother’s household had to be in quite the state of upheaval. Marguerite’s father had recently died, leaving his wife with ten stair-step children, including a one-year-old.
By the 1678 census, Marguerite Hebert was married to Jacques LePrince and was shown with 1 child, a girl, but no age is given. Ages are provided for children in other families in that census.
It’s possible, based on how far upriver Marguerite and Jacques are living, that the census taker simply didn’t venture out that far and asked the closest neighbor that they did visit, or maybe someone at the church in Port Royal.
So, if Marguerite is married to Jacques and has a child in 1678, and IF that child is the first child born after their wedding, then they would have been married by 1676 or 1677.
If this was not their firstborn child, they could have been married prior to 1671 – except that begs the question of where Jacques LePrince was in the 1671 census, and why Marguerite is shown as married, but not in another household with her spouse. Not to mention, where are the children born between 1671 and 1678, aside from that one girl?
If Jacques is living with Marguerite in her mother’s home, which would seem like a reasonable scenario given that her mother was recently widowed and would desperately need help, why isn’t he listed there?
So many unanswered questions!
Who is Jacques LePrince?
Jacques LePrince is a conundrum, too. I wrote about Jacques, here, including the uncertainty about his early years.
To summarize, there is a soldier with the Carignan Company who served in Trois Rivieres, near Quebec City, between 1665 and 1667. Additionally, a man by that same name is found as a valet to a notary in the 1666 Quebec census in Trois Rivieres.
Are those two different men, or one?
Additionally, the Jacques LePrince who is a soldier witnesses a marriage in 1667, and he is probably the same man who witnessed another marriage on April 15, 1671. He is never found in Quebec records again.
Given that the 1671 Acadian census was taken in the spring of 1671, how would it be possible for him to be married in Port Royal, and in Quebec, hundreds of miles away from his wife? That journey takes from 1-3 months by ship.
Furthermore, the transfer of control of Acadia from the English to the French had only taken place in September of 1670, which is when the census was ordered, so it would be very unlikely for Jacques to have been in Acadia before that occurred.
More questions!
Who Is Marguerite Married To in 1671?
It would be outside any norm, but somehow, it might have been possible for Marguerite Hebert to be married to Jacques LePrince in 1671.
It’s also possible that Marguerite could have married, then become widowed prior to the census and was actually living with her mother. If Marguerite was widnowed, would she have been listed as such if she was living with her mother? We don’t know.
Acadian females tended to marry quite young, so she could well have given birth to a child or two by this time. Those children could have died too.
I realize there are a lot of speculative words of uncertainty here – but it’s a perplexing situation, and we can only work with the information we have. The only things we know for sure are her age, that she was or had been married, and that there’s no sign of a husband or household elsewhere. This is not a “normal” situation.
It’s also possible that the Jacques LePrince(s) in Quebec in 1666 and in April of 1671, and the Jacques LePrince who arrived in Acadia at some point and married Marguerite Hebert are entirely different people. The ship L’Oranger sailed from La Rochelle in the spring of 1671 with 50 new settlers to resupply the colony. Jacques could have arrived on that (or another) ship.
Marguerite Hebert’s Children
I’m going to create a table to track Marguerite Hebert’s children, because they aren’t straightforward either.
Nothing about Marguerite is straightforward.
In the 1678 census, based on the neighbors, Jacques La Prence and Marguerite are living far upriver, across from present-day Bridgetown, on four arpents of land, with five cattle, and one female child. Oliver Daigle lives next door, and Marguerite’s brother, Jean Hebert is an unmarried neighbor as well.
The 1686 census shows that Jacques and Marguerite have four children, but does NOT give names and ages, although the census taker does provide that information for other families. They have five sheep and 3 hogs, but no land is listed, nor a gun. Both are quite unusual, especially given that they are literally living the furthest distance from Port Royal of any family in the river valley.
| |
1678 census |
1686 census |
1693 census |
1715 census |
| Jacques LePrince |
No age |
40 (1646) |
Deceased 1691-1693 |
|
| Marguerite Hebert |
No age |
35 (1651) |
40 (1653) |
|
| 1678 |
Girl born in 1678 or before |
Child |
Marguerite, 15 (1678) |
|
| 1680 |
|
Child |
Francois, twin, 13 (1680) |
Francois (no age) |
| 1680 |
|
Child |
Jacques (actually Antoine), twin, 13 (1680) |
Antoine (no age) |
| 1682 |
|
Child |
Missing |
|
| 1684 |
|
|
|
|
| 1686 |
|
|
|
|
| 1687 |
|
|
Estienne, 5 (1687) |
|
| 1689 |
|
|
|
|
| 1691 |
|
|
|
|
| 1692 |
|
|
Francoise, 1 (1692) |
|
By the 1693 census, Marguerite and Jacques have moved from Port Royal to Minas, where, finally, names and ages of children are given. However, Jacques has died, and no land or livestock was listed.
Unfortunately, problems exist with this census accounting too. According to multiple later records, the twin recorded as Jacques is actually named Antoine. Also, we know that son Jean LePrince, who is unlisted here, was born around 1692, so either the child listed as Francoise is actually Jean, or Jean was born to Marguerite in 1693 after the census was taken.
Or, some other even more unusual perturbation has occurred.
Unfortunately, Marguerite now finds herself in the same position as her mother in 1671 – a widow with young children. Except it looks like Marguerite had absolutely no resources – no son-in-law living next door, no land and not even a cow. What was she to do? How was she supposed to feed her family?
How did she feed her family?
In 1671, there were a total of 68 households and about 378 residents in Port Royal and living along the Riviere Dauphin, now the Annapolis River Valley.

In 1693, just 22 years later, in the entire Minas Basin, a much more dispersed area, there were 55 households with 307 residents. Minas was not settled until about 1682, so, in just a decade, it had grown to be about 80% as large as the Port Royal area had been in 1671. By 1693, Port Royal and the surrounding area had about 81 homes and around 500 people.
Marguerite is missing entirely from the 1701, 1703, and 1707 census, and she shouldn’t be. We know she was alive and she had to be living someplace. Unfortunately, widows weren’t enumerated in Minas in 1701 and 1703, but they were in 1707, so where was she living?
What happened in 1704 may provides clues.
The 1714 census is simply a list of male heads of household, so Marguerite is not listed. Her twin sons who married in 1712 are present on the Pisiquid list, as is her son-in-law.
What Do We Actually Know About Marguerite Hebert?

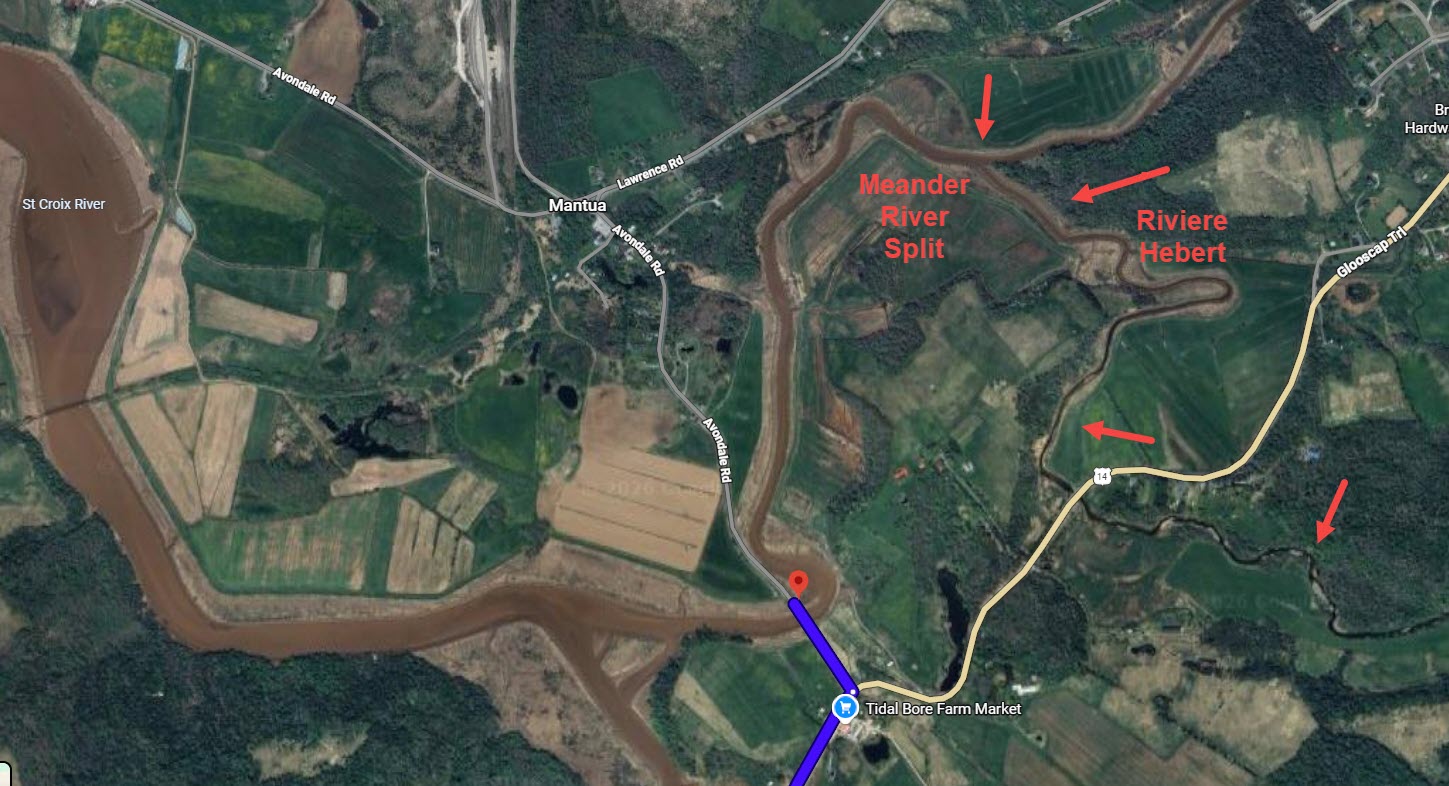
Marguerite’s parents, Etienne Hebert and Marie Gaudet, lived on the upper reaches of the Riviere Dauphin, now the Annapolis River, near today’s Bridgetown. At that time, Bridgetown was called Gaudet Village.

Based on later maps and information, it appears that Marguerite may have grown up in or near the area of Chapman Avenue above the cemeteries. The Old Pioneer Cemetery is shown at far left, above.

Various homes in the Gaudet Village may have been located near the arrows.

Today, we can’t see much from the road at the location of the upper arrow, and I couldn’t get to the land at the lower arrow at all.

The land cross the road from the entrance to the main Riverside Cemetery.

Regardless of exactly where Marguerite’s parents lived, it wasn’t far between the Hebert and Gaudet family clusters along the river. Jean Prince (LePrince) eventually farmed his father’s land across from the Gaudet Village.
Marguerite grew up in a mostly peaceful time, that is, until 1690 when the English brutally attacked Port Royal. Thankfully for Marguerite, she and her family were living upriver, and while they would have heard about the attack, and the subsequent pirate attack, they were beyond the immediate brutality. They may have gone into hiding for safety. Marguerite’s husband, Jacques LePrince, was not on the list of men that were sequestered against their will in the church in Port Royal and forced to sign a loyalty oath to the English monarchy.
However, Marguerite and Jacques may have already left for Minas, or Pisiquid, wherever they originally settled – or these 1690 events at Port Royal may have pushed them over the edge.
Family Migration

Several of Marguerite’s siblings had migrated, or would, to Minas. The family was already used to living remotely from Port Royal, so living even more distantly didn’t much matter. It would have been safer, away from English interference, and provided the advantage of opening new lands that may have been more plentiful and fertile.
Minas soon became the breadbasket of Acadia.
Marguerite’s siblings were:
- Marie Hebert, who was born about 1651, married Michel Le Forest and died about 1677 in Port Royal. Her son, Michel Forest was in Minas by 1693, as was his brother, Pierre. Their sister, Gabrielle Forest married Pierre Brassard and they made their way to Minas too.
- Emmanuel Hebert, born about 1653, married Andree Brun. They lived most of their lives in Port Royal, but after her death in 1727, Emmanuel made his way to Grand Pre where most of his children were already living. He died there in 1744.
- Etienne Hebert, born about 1654, married Jeanne Comeau and was settled in Les Mines by April of 1682 when his second child was baptized. He was one of the founding families and died there in 1713.
- Jean Hebert, born about 1659, married Jeanne Doiron around 1692 in Minas. They settled in Pisiquid. He died around 1744.
- Francoise Hebert, born about 1661, married Jean Comeau. In 1705, the family was still in Port Royal, but by 1707, they were recorded in Les Mines. However, she died in Port Royal in 1713 and was buried in the Saint-Laurent Chapel at BelleIsle.
- Catherine Hebert, born about 1662 married Philippe Pinet. In 1682, they were in Beaubassin, but in 1683, they baptized a child in Grand Pre where they settled. Philippe died about 1710, and by 1714 she had moved to Ile Royale (Cape Breton) with her children where she worked as a gardener. She died in Louisbourg in 1727.
- Martine Hebert, born about 1665, married Nicolas Barrieau. In 1686, the couple was still in Port Royal, but by 1693, they had made the move to Les Mines where she died sometime after 1726, probably in Pisiquid where she was living in 1714.
- Michel Hebert was born about 1666 and married Isabelle Pellerin around 1692. The young couple went to Minas where they were living in 1693. They moved around a bit, were at Riviere des Gaspereau in 1701, back to Les Mines in 1703 and 1707, Port Royal in 1714, then back to Grand Pre/Les Mines in 1715. He died in Les Mines in 1735 or 1736 (date transcription discrepancy) and was buried in the cemetery at Les Minas.
- Antoine Hebert, born about 1670, married twice and lived his life in Port Royal, dying before July of 1753.
Here’s the Shocker!
- Marguerite’s half-sister, Marie Gareau, born about 1677, married twice, but had no children with her first husband. She married the second time to Jerome Darois in 1698. By 1701, they were in Les Mines. In 1706, their son was born as the couple was held hostage in Boston. After their release, they returned to the Minas Basin where Jerome died in 1750. In 1755, Marie was exiled to Virginia where she died.
Wait! What? Held Hostage?
What is going on?
Take a deep breath, pull up a chair and sit down! Maybe get a beverage too.

As it turned out, during Queen Anne’s War, the English, with 17 ships and 550 men, attacked Grand Pre in June of 1704.

By By Hezekiah Butterworth – [https://archive.org/stream/zigzagjourneys00buttiala#page/181/mode/1up Zigzag journeys in Acadia and New France.A summer’s journey of the Zigzag Club through the historic fields of the early French settlements of America.By Hezekiah Butterworth, 1885], Public Domain, https://commons.wikimedia.org/w/index.php?curid=31146683
Their goal was retaliation and to capture Acadian prisoners to exchange for hostages being held by the French in Montreal resulting from the
February raid on Deerfield, Massachusetts. Colonel Benjamin Church’s orders were specifically to:
Use all possible methods for the burning and destroying of the enemies houses and breaking the dams of their corn grounds, and make what other spoil you can upon them.
After arriving in the waters at Grand Pre, Col. Church sent one of his lieutenants ahead, under the white flag of truce, with this proclamation, giving the Acadians and Mi’kmaq one hour to surrender.
We do also declare, that we have already made some beginnings of killing and scalping some Canada men, which we have not been wont to do or allow, and are now come with a great number of English and Indians, all volunteers, with resolutions to subdue you, and make you sensible of your cruelties to us, by treating you after the same manner.
Church expected that his men would have reached the village from behind by the time the hour had passed, but that’s not what happened.
Church’s raid on Grand Pre began something like the Keystone Cops, as his soldiers didn’t understand the extremely high tides in the basin, over 20 feet in some places, with high tide sometimes reaching 52 feet.
Their ignorance combined with the strong tidal rivers caused them to get themselves stuck. At the time they were attempting to come ashore, the tide was receding, which means it was rushing out at more than 33 feet a minute at a rate of about 10 miles an hour, eventually miring his struggling soldiers in mud flats.
I’d laugh if this hadn’t evolved so tragically.
The Acadians were clearly not surrendering, and the delay provided them with the opportunity to evacuate, taking at least some of their cattle and “best goods,” according to Church.
After becoming hopelessly stuck and very muddy, the soldiers extracted themselves and returned to their boats.
The English waited overnight for high tide, but that tide was so high that it elevated their whale boats enough that it exposed them to gunfire from the local militia, Acadian men and their Mi’kmaq allies who had taken cover in the woods along the banks. While the Acadians clearly understood the gravity of the situation, and how vulnerable they were in the face of warships, they must have been at least a little amused. Right up until Church fired his cannon at them, killing one and injuring others, forcing their retreat.
Grand Pre had no fort or defensive structures, so the Acadians were entirely exposed.
Overnight, the English successfully regrouped and tricked the Acadians into believing they were retreating, but they weren’t.
On the morning of the third day, the English attacked the village where, that evening, they proceeded to burn at least 60 homes and even more barns, six mills, and the church. Benjamin Church stated that, “the whole town seemed to be on fire all at once,” and all but one home was burned.
Church’s soldiers tore down the dykes to flood their fields with saltwater, and burned the crops, destroying the harvest.
On the fourth day, Church’s men proceeded on upriver to raid Pisiquid where they took 45 prisoners, then began the trip back to Port Royal to rejoin the fleet blockading the port there.
For comparison purposes, the 1703 Acadian census at Les Mines, which included Pisiquid, shows a total of 61 homes, so Church may have been exactly right – only one house was left standing. That means that Marguerite’s home, along with those of her siblings and children, were all burned.
Another source says that 100 hostages were taken, that the English took as many as they wanted, and that only 5 homes were left standing in Grand Pre. That statement is believed to apply to the larger area surrounding Grand Pre, which would include Pisiquid.
That may have been an slight exaggeration or a bit of braggadocio, given that it was Church’s later telling, but who knows what actually happened. Exaggeration or not, one home or five left standing, the attack was shocking and brutal, and no one in the Minas area was unscathed.
Hostages
The hostages were loaded up and shipped to Boston.
After arriving in Boston, initially the captives were given free rein of the town, but when citizens complained later in 1704, they were confined to Castle William in the harbour and Fort Hill in Boston proper. They were ultimately exchanged in two groups in 1705 and 1706.

Eighteen months later, in December of 1705, the first group of 57 Acadian hostages was released. In September 1706, after being held for 27 months, the second group of 51 was released and arrived back in Port Royal on September 16th.
Thankfully, the parish records at Port Royal reveal a little more of their story. The captives returned to Port Royal before continuing on their way back to Minas. We know this because two baptisms took place for children who were born while their parents were held hostage in Boston. In one case, the parents of the Doiron baby were officially married in the Catholic faith three days later by the priest, although they had been civilly married in Boston during their captivity.
Father Justimen Durand officially baptized the two babies born in Boston just five days after they arrived in Port Royal.

On September 21, 1706 he baptized Louis Mathieu Douairon who was born on February 1, 1706 in Boston to Marie Henry and Noel Douairon (Doiron), and was baptized there by Jean Douiron. Jean had to be either the father or brother of Noel, because there were no other candidates by that name. Noel’s father also lived in Pisiquid, as did his brother.
Of course, this tidbit also tells us that Jean Doiron was being held in Boston at that time as well. It also raises another question. I suspect that there were more than two babies born and baptized in Boston. Why are there only two baptisms in Port Royal? Noel’s father, Jean Doiron, was having children in 1705 and 1706, so I would have expected at least one baptism back in Port Royal – but there are none other than these two, both for children purportedly born on the same day.

On the same page, we find the baptism of Paul Davonois, born February 1, 1706 to Marie Gavoche and Jerome Davonais. Godparent, Pierre Prince.
The names were mistranslated on the archives website and are actually Jerome Darois and Marie Gareau
On the twenty-sixth of September 1706, I the undersigned, performing the curial functions, supplied the ceremonies of baptism for Paul Darois, son of Jérôme Darois and Marie Gareau, his lawful wife.
He was born in Boston on the first of February of the present year, and was baptized conditionally.
The godfather Jean Prince was not able to be present, and the godmother…
Signed:
Fr. Justimen Durand, Recollect missionary
Marie Gareau was Marguerite Hebert’s half-sister. Jean Prince (LePrince) was Marguerite Hebert’s son, which means Jean was Marie Gareau’s half-nephew. Jean was not at the baptism, which is somewhat remarkable because he married at Port Royal in 1715, which suggested the possibility that he grew up there.
If Jean was not in Port Royal in 1706, when he was 13 or 14, that hints that he may have been living with his mother in Pisiquid, and that his mother, Marguerite, if she was a hostage, was not in the group released in 1706 either. Of course, that doesn’t mean she wasn’t kidnapped. She, along with her family, could have been released in 1705.
It’s unusual that the only two babies baptized were born on the same day. Perhaps that date was an estimate.
The Acadians in Grand Pre had clearly not been prepared to defend themselves. Needless to say, they never again trusted the English – and with good reason. Fifty years later, the English did even worse to the Acadians.
If 108 hostages were taken, I wonder how many residents were not taken.
In 1707, the year after the last of the hostages had been returned, there were 88 households in all of Minas with about 575 residents. That means that about one in five people across the entire region were taken hostage.
In 1714, the only census that lists Pisiquid separately, there were 57 households, with 372 people. Realizing that 1714 is a decade later, it looks like about one third of the population of Pisiquid was taken hostage.
In 1714, there were an additional 47 households listed separately at Minas, so the region-wide population probably hadn’t changed much.
My initial reaction was that the English probably didn’t want children, so most of the hostages would have been couples or at least not young children. However, further research revealed that exactly the opposite was true. According to Church’s own memoirs, the majority of the hostages were wives and children of Acadian farmers. He stated that this was a tactical decision, and said he took between 45 and 100 prisoners at Grand Pre, a large portion of whom were families.
I can’t help but wonder if the Pisiquid families weren’t warned in enough time to disappear into the woods or seek safety elsewhere. Surely, they didn’t just sit there, waiting.
Maybe they thought that one of the safest places they could be was far down the Riviere Hebere, where the English wouldn’t have dared to go. The English had never attacked there before, so maybe they were well-enough hidden? That’s initially what I thought too, but that clearly wasn’t true.
Where was our Marguerite? Was she taken hostage? Her children? She would have been 52 or 53 at the time.
In 1704 when the English arrived, her youngest child would have been Jean LePrince who was about 11 if he was born after the 1693 census. Francoise, if she actually existed and was still living was 12. Estienne would have been about 15. The twin boys, Francois and Antoine, 22.
There’s a lot of confusion about Marguerite’s oldest daughter or two oldest daughters, so I’ll leave that for the next section.
At least Marguerite and her siblings had each other. Someone cared for her sister, Marie Gareau’s children during her two-year absence, assuming they weren’t all taken hostage too.
Pisiquid
Only one of Marguerite’s siblings remained permanently in Port Royal, aside from her older sister, Marie who died there in 1677, although Marie’s husband and children made their way to Minas as well.
Virtually all of Marguerite’s family left Port Royal, so it’s little wonder that she and Jacques LePrince departed too. This is presuming that Jacques didn’t die in Port Royal first.
Marguerite couldn’t have been in a better place to receive the support she needed while raising her family alone. Still, I wonder what happened to her, how she lived, where and with whom? I also wonder if she remarried and we simply don’t have the records, meaning we don’t know to whom. It was quite unusual for widows, especially with young children, to remain single. That would also explain why she doesn’t appear in later censuses where she would be expected to be found.
I did have one other fleeting thought. I hope that Jacques LePrince didn’t just disappear, either willingly or against his will – leaving Marguerite with no closure or resolution, and also unable to remarry.
Sometimes the frontier was unforgivingly harsh.
Marguerite’s Children
We have yet another mystery.
The census tells us only so much.
We already know the census was “odd,” with no information provided when other families had information, and then in another census listing erroneous names of children.
I don’t want to read anything more into this than there is, but there are a lot of “strange” things, both with Jacques LePrince and his family, and then with their son, Jean LePrince and his.
Some of the circumstances leave me uneasy.
That said, let’s see what we have.
Some of the children are relatively straightforward, but the most challenging ones are the oldest.
I’m going to discuss Marguerite’s children in birth order.
Referring back to the chart I made for her children, you’ll recall that Marguerite was noted as age 19 and married in the 1671 census. However, no husband or family is recorded, aside from her deceased father, mother and siblings.
An Acadian female would be expected to have married by age 19, so it would be somewhat unusual if Marguerite had NOT married.
If we are to take the census-taker at his word, this would mean one of three things:
- Marguerite’s husband and household were missed in the census
- Marguerite was married, but had been widowed, and was living with her mother who had recently been widowed and had young children.
- Marguerite was married or widowed, and had given birth to children or was pregnant. In either case, there are no signs of multiple children in the next census, so we are left to presume Marguerite buried three or four children between 1671 and 1677, or so.

Given that Marguerite and family were living in the proximity of Port Royal, those babies might have been buried beside the church in Port Royal, which isn’t terribly likely since they lived far upriver.

They could have been buried at Saint Laurent “Mass House” at BelleIsle, which is closer to where they lived and where one of Marguerite’s siblings is buried.

Or, perhaps they were laid to rest in the old pioneer cemetery in what was the Gaudet Village, found in present-day Bridgetown. Marguerite’s mother was Marie Gaudet, so the unmarked portion of the Old Pioneer Cemetery would be my best guess, but we’ll never know for sure.

Or, maybe the original cemetery was located in the oldest part of the Riverview Cemetery, off of Chapman Avenue, in the unmarked portion near the river.

We don’t know which Bridgetown cemetery is older and was established first, and no records exist in any of the churches for that timeframe.
One thing is certain. It would have been virtually impossible to travel all the way back to Port Royal for burials, and burials couldn’t exactly wait. Out of pure necessity, there was certainly a cemetery someplace here and it was likely continued when the New England settlers took over the Acadian farms after 1755.
Marguerite LePrince
By the 1678 census, Marguerite is shown living with her husband and one female child, with no age given. In 1686, four children are reported, but no names or ages are included. It’s not until 1693 that we have names for Marguerite’s children.
Therein lies the rub. We know the 1693 census wrongly recorded at least one of Marguerite’s children, and possibly two or three. That’s not very good odds and reduces our confidence in the remainder of the information.
In 1693, Marguerite Hebert, the widow of Jacques LePrince, is living in Mines.
The 1693 census shows that Marguerite’s oldest daughter, also named Marguerite, is age 15, which maps exactly to the one female child in 1678. Is Marguerite that child?
However, there’s one Marguerite LaPrincesse, age 12, living with Daniel LeBlanc in Port Royal at BelleIsle, as a servant. Daniel LeBlanc is 66, living with Francoise Godet, his wife, 76, Pierre 28, Pierre the son of Pierre, 7, and two servants, Jean LaForet and Marguerite LaPrincess. Daniel has a large farm with 20 cattle, 35 sheep, 9 pigs, on 18 arpents of land, and owns 3 guns. He is clearly not a poor farmer.
Given that Daniel’s son, Joseph LeBlanc, seems to be widowed and raising a child, they may have taken servants to help with both household and farm chores.
We don’t know when Jacques LePrince died, or when Marguerite Hebert moved to Mines. Regardless, her daughter, Marguerite LePrince, can’t be living in two places at the same time.
There are also no other Prince males in Acadia, so if the Marguerite LaPrincess living with Daniel is a Prince or LePrince, she has to be Jacques’ child.
I have to ask this out loud. Why on earth would a mother leave her 12-year-old daughter as a servant in someone’s home and move to the next frontier?
The only reason I can imagine is that Marguerite Hebert was just dirt poor and had no other choice. That’s certainly possible following Jacques’ death. Yet, she had several siblings in Minas who would have helped feed her, so I’m baffled.
Marguerite LePrince married Francois Tillard around 1701 in Port Royal. My first thought was that this was very unlikely to be Marguerite Hebert’s daughter, but a deeper dive shows that it almost certainly was.
Marguerite LePrince and Francois Tillard had four children:
- Marie Louise Tillard born around 1701 and died in 1751. Marie married Claude Trahan of Pisiquit about 1724.
- Francois Tillard born 1712 in Annapolis Royal, married Marie Denis in 1732 in Minas. His father is Francois Pillard, decd, and Marguerite Prince of Pigiguit of the parish of l’Assomption de la tres Ste. Vierge. Antoine LePrince (Marguerite Hebert’s son) is a witness. In 1742, Rene LePrince is Godfather to their child. L’Assomption is the Pisiquid parish, and the one closest to Riviere Hebere.
- Marguerite Tillard was born in 1714 in Port Royal, and married in Minas in 1734 to Joseph Dueron from Cobequid. Her mother is Marguerite LePrince.
- Magdelaine Tillard was born about about 1717 and married in 1738 in Minas to Francois Doiron. Mother is listed as Marguerite LePrince.
Marguerite LePrince apparently moved to Minas after Francois Tillard died, sometime after 1717 and before 1724 when her daughter, Marie married Claude Trahan from Pisiquid.
In 1734, Marguerite LePrince, daughter of Jacques LePrince and Marguerite Hebert, both deceased, received dispensation for a third degree of consanguinity and married in Minas to Jean Hebert, who lived at Cobequid. Their common 3rd degree ancestor would have been the unknown parent of Marguerite Hebert’s father, Etienne, and his brother, Antoine Hebert. This confirms that Marguerite LePrince who married Jean Hebert is Marguerite Hebert’s daughter.
All of this, taken together, strongly suggests that indeed, the Marguerite LePrince who was a servant to Daniel LeBlanc in 1693, and who was also listed with her mother in Minas, is the same person who remained in Port Royal when her mother moved.
What we don’t know is whether Marguerite was born in 1678, as the census with her mother states, or was born in 1681 as her age listed with Daniel LeBlanc states.
We don’t know when Marguerite LePrince died, other than it was after her daughter Magdelaine Tillard married in 1738.
This brings us to the second challenge. An unrecorded daughter.
Anne LePrince
In the 1678 census, only one child is shown for Marguerite Hebert, a daughter. In 1686, four children are shown, so one born around 1678ish, the twins in 1680, and a child in the 1682 slot that’s missing later.
Or, maybe, just maybe, the oldest child is actually Anne LePrince, followed by the twin boys, then Marguerite, born in 1681.
If that is the case, then where is Anne in 1693?
We already know that the 1693 census is flawed. Maybe Marguerite actually was living with Daniel LeBlanc and never moved to Minas with her mother.
Perhaps the “slot” for Anne is taken by Marguerite’s name. We know that the census-taker got one of the twins’ names wrong too, and possibly misreported Francoise in place of Jean.
So, the next question is, how do we know Anne existed, at all?
- On July 18, 1709, Rose Rivet was born and baptized that September in Minas to Etienne Rivet and Anne Prince.
- Two years later, in the same months, Michel Rivet was born and baptized in Minas to the same parents. Antoine Prince, Marguerite’s son, stood as Godfather.
This suggests that Anne LePrince was married to Etienne Rivet by at least 1708. The Minas parish records only exist from 1707. If Anne was born in 1678, she would have been 30 by this time, a spinster by Acadian norms. Of course, she could easily have been married several years earlier.
There’s more.

After the Expulsion of 1755, some of Anne’s descendants wound up on Belle-Ile-en-Mer where they were initially housed in a warehouse adjacent the Palais inside one of the city gates where they gave depositions about their French origins.

While that sounds brutal, it wasn’t, because for the first time in more than a decade since the 1755 Expulsion, they were safe and free and awaiting assignment to land, homes and livestock in various locations around the island.

When I first tried to sort through this quagmire of testimony, I initially thought that this Anne was not the daughter of our Marguerite Hebert, but putting everything together, it certainly appears that she was.

In 1755, the Acadians who eventually wound up on Belle-Ile-en-Mer were rounded up, held in Fort Edward at Pisiquid where they were informed of their fate, then shipped to Virginia, where their ship was ultimately refused. The ship was sent on to England, where the Acadian passengers suffered, essentially as prisoners of war, living in cramped conditions in warehouses along the docks until after the Treaty of 1763 ended the conflict. 242 Acadians arrived in Liverpool, but many died of disease, especially smallpox during the seven years they were held there. Many of the English complimented the work ethic of the “Neutrals.”

In 1763, with the end of the war, they were repatriated to various locations in France, where the French government and citizens helped them establish new homes.

One of those locations was Belle-Ile-en-Mer.
Part of the process was to explain how they were French, so depositions were taken where we learn the following:
- Rose Rivet, born in 1760, was the child of Jean Rivet and Rosalie Bonniere. Jean Rivet was the son of Estienne Rivet and Anne LePrince who lived at Pisiquid and were members of the Sainte Famille Parish in 1727.
- Anne’s first child, Marie Rose Rivet swore that she was born at Pisiguit, in the parish of La Sainte Famille, on July 18, 1707 to Anne LePrince and Estienne Rivet, and that Anne (her mother) died in Maryland after she was deported in 1755.
The deposition then continues to say that all of Anne LePrince’s children were born at Pisiquit, when they were born, and what happened to them.
- Michel Rivet born in 1709 died in 1740.
- Etienne Rivet born in 1717 was transported to Maryland with his family.
- Anne Rivet born in 1719 died in Pisiguit in 1750.
- Claire Rivet born in 1723 was transported with her family to Maryland
If Claire was Anne’s youngest child, Anne would have been 43 years old when Claire was born, which is just about right.
Anne LePrince survived the deportation itself, and died, hopefully surrounded by her family, in Maryland. She would have been 77 in 1755 when forcibly herded onto those ships, with no possessions, to endure the freezing winter crossing.
Francois LePrince
Francois is the only one of Marguerite Hebert’s children whose documentation is consistent.
Twins were rare in Acadia. He and his twin brother, Antoine, were born about 1680 in Port Royal, according to the census.
By 1693, he was age 13, living with his widowed mother in Minas.
Francois and his twin brother had a double wedding in 1712 in Minas. Actually, it was a triple wedding, but I haven’t figured out a blood relationship to the third couple, if they were related. Fortunately, the Minas parish registers were taken to Louisiana and still exist.
- Francois Prince (<father’s name omitted> and Marguerite Hebert) married May 23, 1712 Catherine Benois (Martin Benois and Marie Chosegros of Pigiguit), witnesses Jacques Terriot, signed, Pierre Forets, signed, Guillaume Trahan, mark, Francois Rimbauld, mark, Pierre Benois, mark, Francois Michel, mark, Francois Prince, mark, Catherine Benois, mark. Triple wedding with Louis Sire/Marie Joseph Michel and Francois Prince/Catherine Benois (SGA-1,11)

Based on this and other information, while they were probably married in the church in Minas, they would have been living in Pisiquid, which is a significant distance from where the church was located in Grand Pre, the hub of the settlement.
Francois and Catherine had seven known children, and probably another five or six who did not survive.
We don’t know when Francois died, other than it was after his youngest child was born in 1731, and before the 1750 census on which he does not appear.
Many of his children sought refuge at Port LaJoye on Ile-St-Jean, today’s Charlottetown on Prince Edward Island, which was initially supposed to be a safe-haven. From there, about 3500 Acadian refugees were imprisoned and shipped on to France. Three overcrowded ships, the Duke William, the Violet and the Ruby tragically sank, killing hundreds.
Francois’s youngest son, Claude, perished on December 13, 1758 along with his wife and three children when the Duke William sank after being separated from the rest of the fleet during a severe storm. Her mast broke, and her hull failed, causing the ship to be overwhelmed with water and sink near Falmouth, England.
More than 360 passengers, if you can call them that, drowned, but the captain, crew, priest and a few others managed to survive in small boats.
Noel Doiron, whose son, Louis Mathieu, was born in Boston as a hostage in 1706, was aboard that ship, along with Louis, Noel’s wife and four of their other children – 50 years after they survived being kidnapped, held as hostages, and released.
I can only imagine the indescribable terror.
Antoine LePrince

Antoine LePrince celebrated his wedding with his twin brother, Francois. Perhaps the wild roses that still grace the Acadian homelands were in full bloom.
- Antoine Prince, (Jacques Prince, decd, and Marguerite Hebert, of the parish of des Mines), married May 23, 1712 Anne Trahan (Guillaume Trahan and <mother omitted>, habitants of Piguguit), witnesses Jacques Terriot, signed, Pierre Forets, signed, Guillaume Trahan, mark, Francois Michel, mark, Antoine Prince, mark, Anne Trahan, mark. Triple wedding with Louis Sire/Marie Joseph Michel and Francois Prince/Catherine Benois (SGA-1,11)
I can’t help but think about that joyful gathering. They probably planned carefully, coordinating a trip to Minas so a priest would be present. Everyone from the neighborhood, even though it was miles away, probably made the journey because they wanted to attend all three ceremonies.
Had they married in the local parish, Sainte Famille, at Pisiquid, we would have no record today because those parish records no longer exist.
Antoine LePrince and Anne had nine known children, and probably another four or five who didn’t live to adulthood.
In 1730, Antoine’s name appeared on the list of Pisiquid inhabitants who agreed to swear an oath of allegiance to the English crown. He may have had little choice in the matter.
The Belle-Ile-en-Mer depositions state that his great-great-great-granddaughter died at Liverpool in June of 1763. It further states that Antoine LePrince and his wife both died as Pisiguit.
Antoine’s Daughter, Anne LePrince – Marguerite’s Granddaughter
Additionally, the depositions tell us that Anne LePrince, daughter of Antoine, married Sylvain LeBlanc who died in Liverpool in 1756, but “Anne LePrince is living in Morlaix with her family.”

Anne was born about 1724, and was listed on the ship manifest for “The Sturgeon,” bound for France from Liverpool, with her children:
- Anastasie LeBlanc
- Joseph LeBlanc
- Marguerite LeBlanc
- Modeste LeBlanc became a Carmelite nun at Morlaix, taking the name of Sister Augustin de Saint-Francois de Sales, according to Karen Theriot Reader, here
Anne LePrince lived in the port city of Morlaix for the last 20 years of her life, until 1794. She is listed on the census there in 1772, 1786 and 1792 and was among the city’s poor. She and her daughter reportedly sold knitting to help support themselves.
According to the historian Clarence J. d’Entremont, Anne was executed by guillotine along with one of her daughters Anastasie (others say it was Angelique, but there’s no evidence of a daughter by that name), and Abbé Clech, a priest, on July1, 1794 in Brest, France.

Anne lived here, at 97 Rue des Vignes, in Morlaix, with her daughter or daughters.

It was here that she and her daughter sheltered the priest who arrived at their door asking for a place to stay during the French Revolution – for which they were taken to Brest, tried, and subsequently beheaded.
They were arrested on June 21st, 1794 in Morlaix and taken to Brest on the 23rd.

They were all three imprisoned at the castle, with Anne and her daughter accused of having harbored the priest in their home, thus defying the laws of the Revolution.
They would have been led down these stairs into the castle dungeon.

On July 1st, their trial began at 8 in the morning, here, in the Revolutionary Tribunal Chapel, the site of the former Jesuit seminary, at today’s intersection of Rue de Lyon and Rue Duquesne in Brest. These buildings no longer exist. If not torn down before, Brest was heavily bombed during WWII.
The verdict was pronounced nearly immediately, which said:
The Tribunal ordains that the said August Clec’h shall be delivered to the executor of the criminal cases to be put to death according to articles ten (etc.). Sentences Anne LePrince, Anastasie LeBlanc, her daughter, to die according to articles 2 and 3 (etc.)
Executions were public spectacles, designed to take place where the maximum number of people could gather and bear witness. They took place nearly daily during this period in history, referred to as the Reign of Terror, at Place du Chateau, in front of the castle.

Anne, her daughter and the priest, all three condemned, would have been marched through the streets immediately thereafter, if not carried by cart, as part of the exploitive “entertainment”.

The locations shown on this early map no longer exist as they did at that time. The Place du Chateau functioned as both a military esplanade and site of public executions.
The crowd would have gathered. Who was going to be guillotined today? What would they do? Beg? Cry? Confess?
At noon, first the priest, then Anne LePrince, by then an elderly woman of at least 70, followed by her daughter, Anastasie, about 38 or 39, climbed onto the scaffold platform to provide the day’s amusement for the gathered crowds by having their lifeblood spilled.
I can’t even begin to imagine watching this horrible fate happen to YOUR MOTHER!
I can’t even look at a guillotine without becoming nauseous, so you’ll have to google that one for yourselves.
After the Revolution ended, public executions slowed and ended at this location in 1839. In the 1840s, France began “civilizing” former sites with such a brutal history into promenades and public squares.

In many cases, what was originally the “central figure”, meaning the scaffold and execution site, was replaced with a fountain, also located in the center of the same expansive location.
So it was probably right here that Anne and her daughter were murdered. Not for violence, or a crime – but for refusing to surrender their faith and giving a man of the cloth shelter in their home.
Blood was replaced by water. Gardens and fountains softened the space, their gentle sounds drowning out the echoes of terror that once filled the square. Gone were the sounds of the crowd as they watched. Did they cheer? Gasp? Did they turn away? This was sport.
People forgot – or chose not to remember – what happened here. Over time, the cheerful voices of the next generations wafted in the air where screams once echoed, replacing the intentionally-erased and deliberately-buried trauma of the place where bloodthirsty crowds watched hundreds die beneath the executioner’s blade – one terrified human being at a time.
Among them stood Marguerite’s granddaughter and her child, condemned not just for what they had done, following the example taught in the Bible, but for what they refused to deny.
I can’t even begin to comprehend Anne’s bravery. She assuredly knew the risks.
For that, she was martyred.

By 1902, just 108 years later, La Place du Chateau had become a gardens for families as pictured in this post card. No one knew what had systematically occurred here.

By 1944, this entire area had been bombed into oblivion during the siege of Brest.
In a different location, but for the same underlying reason, 32 Carmelite nuns were executed. They refused to take the required oath and revoke their vows, so they were loaded onto carts and went to their death singing hymns before being publicly executed and buried in a mass grave.
This sends shivers up my spine. I hope that Anne’s other daughter, Modeste, didn’t suffer this fate. Given that she was a nun in Morlaix, she assuredly knew what happened to her mother and sister.
While Anne and her daughter, Anastasia, must have been utterly terrified, I picture them defiantly singing as well. They were already convicted and sentenced to death. What else was left to do?
After their execution, their bodies and heads would have been thrown into the cart, which would have been drawn through the city to some obscure trench on the outskirts, leaving a trail of blood behind. Clearly, there was no family to claim them, and no one would risk claiming a priest.
There is some great irony knowing that because of the unique circumstances, Anne and her daughter unquestionably received last rites.
You can read the rest of Anne’s story, here and here.
I hope Anne’s grandmother, Marguerite, was watching over her and waiting on the other side to embrace and greet Anne and Anastasia. Maybe one day we will have two official martyrs in the family.
Anne wasn’t alone in Morlaix, at least not initially. Her sister, Marguerite was with Anne in Virginia, then in Liverpool, then Morlaix, but died in 1778.
Two of Anne’s siblings made it to Pennsylvania, one landed in Maryland, one eventually made their way to Quebec, another died in Liverpool, and one, Tranquille, died in Louisiana in 1798.
Antoine LePrince’s children were truly scattered to the winds.
Marguerite’s Missing Children
After Francois and Antoine LePrince’s births about 1780, and probably Marguerite LePrince’s birth around 1781, there are at least two missing children, in 1783 and 1785.

They died before the 1786 census, and like the others, they are probably buried at either St. Laurent, above, where nothing remains today, or one of the two cemeteries in Bridgetown.
Estienne LePrince
Estienne is shown in 1693 as five years old, which means he would have been born about 1687.
Unfortunately, Estienne apparently died before marrying, because we have no further records. He would have married in the 17-teens, meaning he or his children would have probably appeared in some record.

Estienne is probably buried either in the cemetery at Grand Pre, marked today by this cross beside the church, at Sainte-Famille, or maybe in a family chapel cemetery along the Riviere Hebere.
More Missing Children
Following Estienne in the census are spaces for children who would have been born in 1689 and 1691.
Two more small graves for Marguerite. Two more babies for her to prepare for burial.
This was also the time period in which Marguerite’s husband, Jacques LePrince, died as well.
They were all three gone by the 1693 census.
Poor Marguerite. Her aching heart!
Francoise LePrince
We really have no idea if Francoise actually existed, or if the census-taker was falling down on the job again.
In the 1693 census, in Minas, Francoise is shown as one year old. If she existed, she perished.
We do know, however, that a male child, Jean LePrince was born about that time.
Jean LePrince
Jean LePrince was either:
- Actually Francoise in the census, and the name was a mistake
- Or he wasn’t living with his mother in 1693
- Or he was born after the 1693 census, and after his father had died
We know beyond any doubt that Jean was the child of Jacques LePrince and Marguerite Hebert.
He married in 1715 in Port Royal, where his parents are listed as Jacques Prince, deceased, and Marguerite Hebert, habitant of Mines.
Jean lived at Port Royal. Now, based on the fact that Marguerite Hebert’s daughter, Marguerite LePrince, who married Francois Tillard around 1704 in Port Royal and seems to have lived a nontrivial portion of her life in Port Royal too, I can’t help but wonder if Jean LePrince never left Port Royal.
Why is this family so split between those two locations? Port Royal and Minas. Two children living and marrying in the Port Royal area after their father died and their mother is found with the rest of their siblings in Minas in 1693. In 1693, both Jean LePrince and Marguerite LePrince would have been young teens.
Maybe someone else took them in to raise, but why?
Maybe Jean LePrince hadn’t yet been born when the census was taken in 1693.
Nothing else gives his age or even a hint at his birth year.
Maybe his mother, Marguerite, couldn’t handle any more than she was already handling.
It has always been presumed that Jean was the child born in 1693, not Francoise. However, in 1715, at what would be age 22 if he was born in 1693, the married a 33-year-old widow. Acadian males typically did not marry that young, and not to someone 11 years older. Now I wonder if Jean was actually one of those older missing children.
If so, then where was he at the time?
Why isn’t he listed in the census?
But then again, why isn’t Anne LePrince listed in the census?
Why aren’t Jacques LePrince and Marguerite Hebert’s children listed with names and ages in 1686 when everyone else’s children have names and ages?
And why was Marguerite “LePrincess” listed in Port Royal as a servant to Daniel LeBlanc in 1693, age 12, and also in Minas with her mother at age 15?
Why was this family so scattered?
Not to mention, even harkening back to 1671, where was Marguerite’s husband, Jacques LePrince?
Something has always been a bit unsettling about this family.
I know there’s a backstory that we will never know.
There are just too many anomalies to be coincidence.
I hope, whatever the circumstances, Marguerite was able to find peace during her earthly existence and joy in her 34 grandchildren.
Marguerite’s Death and Burial

Thanks to marriages recorded by the priest in the St. Charles aux Mines Parish, which is the church at Grand Pre, we know that Marguerite’s twin sons were married in a triple ceremony in 1712, and she was living then. Marguerite is listed as “of the parish of Minas” but both of her sons married women from Pisiquid (Fort Edward on this map), which is highly suggestive of where they actually lived, regardless of where they married. It’s also possible that the priest was visiting locally and they were actually married at Pisiquid, not in Grand Pre.

In 1714, both twin sons are living in Pisiquid as is Marguerite’s daughter, Anne Prince, with her husband, Estienne Rivet. Additionally, several of Margaret’s siblings have settled there too, including her brother Jean Hebert, sister Martine Hebert, with husband Nicolas Barrieau (Barillot), her half-sister, Marie Gareau, who had been held as a hostage with husband Jerome Darois, Marguerite’s deceased sister Marie’s children, plus several nieces and nephews. Not to mention many Forest, Trahan and LeJeune neighbors – all people from the old Port Royal upriver neighborhood, most of whom Marguerite was probably related to one way or another – or maybe even multiple ways.
Pisiquid was very clearly a close-knit family community.
Fifty-six households would assuredly be enough people to warrant a convenient cemetery, perhaps at the “Ville Trahan” family chapel.
In 1715, Marguerite’s son, Jean LePrince, who is entirely omitted from the census, married in Port Royal, and she is listed as living in Minas.
By 1734, when her daughter, Marguerite LePrince, married in Minas, Marguerite was noted as deceased, but there was no death record for her in the parish register between 1712 and 1734. Of course, parish records may not have been complete, or, more likely, she wasn’t buried by the priest in the cemetery beside the church in Grand Pre. Only two Pisiquid burials are recorded in the Minas parish records, which stretch from 1707-1748.
So Marguerite died sometime in the span of 19 years, between 1715 and 1734, probably at Pisiquid, which puts her age between 63 and 82.
While we don’t know when Marguerite departed this life, the one thing we can surmise is that her funeral sermon would have been given either by the visiting priest, later, or a neighbor or family member when she died.

The closest consecrated ground before 1722 was at the Sainte Famille Cemetery on Gabriel Road in Falmouth, established in 1698.
After 1722, the closest cemetery associated with a parish church was at l’Assumption, where Fort Edward stands today. The cemetery there is considered a “lost cemetery,” but archaeologist Jonathan Fowler suggests that the cemetery is located within sight of the remaining blockhouse. You can read his article, here.

Only the block house remains today, but in the above satellite image of the Fort Edward National Historic Site, you can clearly see the outline of the original fort. According to Dr. Fowler’s research, compiled from earlier photos and descriptions of surrounding willows, he believes that the original parish cemetery was located about 200 feet from the original fort, which was built where the church was torn down, in the approximate location of the red circle.
Neither parish location was close or convenient to people living along the Riviere Hebere, another nine miles east of the church at l’Assumption, and both required crossing tidal rivers.

Click any image to enlarge
We know that, in addition to the known parishes, there were two family chapels, one of which, Trahan, was between l’Assumption and the Riviere Hebere, where Marguerite and her children assuredly lived.
The Trahan Village is located someplace near Three Mile Plains, according to the Avon River Heritage site.
Maybe Marguerite is buried there, or maybe the priest consecrated family plots at remote Acadian homesteads, and Marguerite is buried in a long-forgotten Hebert family cemetery. Of course, all Acadian cemeteries were abruptly abandoned in 1755, by no choice of the Acadian families. Perhaps Marguerite was buried in a cemetery later adopted by the English planters who were given the Acadian farms after the 1755 Expulsion.
It’s unlikely that the new inhabitants, New England settlers who took over farming the Acadian homesteads, would have destroyed existing graves. They may have simply begun burying their own family members in an already established graveyard. That’s exactly what happened in Port Royal.
Two cemeteries, the Old Riverview Haven Cemetery and the Old St. James Anglican Cemetery, both with many unmarked graves, are located on Chambers Road.

Chambers, a remote dirt road, inaccessible even on Google maps, runs along the Rivière Héberè, now the Herbert River, just beyond where the tidal bore settles and becomes nothing more than a ripple, whispering secrets from the past. It traces the edge of the marshlands where Marguerite lived for the second half of her life, since at least 1693.
It’s probably along this tranquil stretch, close to her home, that Marguerite spent her final days.
Wherever you are, Marguerite, rest easy.

_____________________________________________________________
Share the Love!
You’re always welcome to forward articles or links to friends and share on social media.
Subscribe!
If you haven’t already subscribed, it’s free. You’ll receive an e-mail whenever I publish by clicking the “follow” button at the top of the main blog page, here.
Help Keep This Blog Free
I receive a small commission when you click a vendor link in my articles and purchase that item. This does NOT increase your price but helps me keep the lights on and this informational blog free for everyone. Please click on the affiliate links in the articles or to the vendors below if you are purchasing products or DNA testing.
Thank you so much.
DNA Purchases and Free Uploads
Genealogy Products and Services
My Books
Genealogy Books
Genealogy Research
Like this:
Like Loading...