
On September 27th, 2018 Family Tree DNA published the largest Y haplotree in the world, based on SNP tests taken by customers. Now, less than two weeks later, they’ve added an exhaustive mitochondrial DNA (mtDNA) public haplotree as well, making this information universally available to everyone.
Family Tree DNA’s mtDNA Haplotree is based on the latest version of the mtDNA Phylotree. The new Family Tree DNA tree includes 5,434 branches derived from more than 150,000 full sequence results from 180+ different countries of origin. Family Tree DNA‘s tree has SIX TIMES more samples than the Phylotree. Furthermore, Family Tree DNA only includes full sequence results, where Phylotree includes partial results.
This new tree is a goldmine! What does it provide that that’s unique? Locations – lots of locations!

The Official Phylotree
Unlike the Y DNA tree, which is literally defined and constructed by the genetic community, new mitochondrial DNA branches cannot be added to the official mitochondrial Phylotree by Family Tree DNA. Haplogroups, meaning new branches in the form of SNPs are added to the Y tree as new SNPs are discovered and inserted into the tree in their proper location. The mitochondrial DNA phylotree can’t be expanded by a vendor in that manner.
The official mitochondrial Phylotree is maintained at www.phylotree.org and is episodically updated. The most recent version was mtDNA tree build 17, published and updated in February 2016. You can view version history here.
Mitochondrial Phylogenic Tree Version 17
Version 17 of the official mitochondrial tree consists of approximately 5,400 nodes, or branches with a total of 24,275 samples uploaded by both private individuals and academic researchers which are then utilized to define haplogroup branches.
Individuals can upload their own full sequence results from Family Tree DNA, but they must be in a specific format. I keep meaning to write detailed instructions about how to submit your full sequence test results, but so far, that has repeatedly slipped off of the schedule. I’ll try to do this soon.
In a nutshell, download your FASTA file from Family Tree DNA and continue with the submission process here. The instructions are below the submission box, so scroll down.
In any case, the way that new branches are added to the phylotree is when enough new results with a specific mutation are submitted and evaluated, the tree will have a new branch added in the next version. That magic number of individuals with the same mutation was 3 in the past, but now that so many more people are testing, I’m not sure if that number holds, or if it should. Spontaneous mutations can and do happen at the same location. The Phylotree branches mean that the haplogroup defining mutations indicate a common ancestor, not de novo separate mutations. That’s why analysis has to be completed on each candidate branch.
How do Mitochondrial DNA Branches Work?
If you are a member of haplogroup J1c2f today, and a certain number of people in that haplogroup have another common mutation, that new mutation may be assigned the designation of 1, as in J1c2f1, where anyone in haplogroup J1c2f who has that mutation will be assigned to J1c2f1.
While the alternating letter/number format is very easy to follow, some problems and challenges do exist with the alternating letter/number haplogroup naming system.
The Name of the Game
The letter number system works fine if not many new branches are added, branches don’t shuffle and if the growth is slow. However, that’s not the case anymore.
If you recall, back in July of 2012, which is equivalent to the genetic dark ages (I know, right), the Y tree was also represented with the same type of letter number terminology used on the mitochondrial tree today.
For example, Y DNA haplogroup R-M269 was known as R1b1a2, and before that the same haplogroup was known as R1b1c. The changes occurred because so many new haplgroups were being discovered that a new sprout wasn’t added from time to time, but entire branches had to be sawed off and either discarded or grafted elsewhere. It became obvious that while the R1b1a2 version was nice, because it was visually obvious that R1b1a2a was just one step below R1b1a2, that long term, that format just wasn’t going to be able to work anymore. New branches weren’t just sprouting, wholesale shuffling was occurring. Believe it or not, we’re still on the frontier of genetic science.
In 2012, the change to the SNP based haplogroup designations was introduced by Family Tree DNA, and adopted within the community.
The ISOGG tree, the only tree that still includes the older letter/number system and creates extended letter number haplogroup names as new SNPs are added provides us with an example of how much the Y tree has grown.

You can see that the letter/number format haplogroups to the far right are 19 locations in length. The assigned SNP or SNPs associated with that haplogroup are shown as well. Those 19-digit haplogroup names are just too unwieldy, and new haplogroups are still being discovered daily.
It’s 2012 All Over Again
That’s where we are with mitochondrial DNA today, but unlike Y DNA naming, a vendor can’t just make that change to a terminal SNP based naming system because all vendors conform to the published Phylotree.
However, in this case, the vendor, Family Tree DNA has more than 6 times the number of full sequence mitochondrial results than the mitochondrial reference model Phylotree. If you look at the haplogroup projects at Family Tree DNA, you’ll notice that (some) administrators routinely group results by a specific mutation that is found within a named haplogroup, meaning that the people with the mutation form a subgroup that they believe is worthy of its own haplogroup subgroup name. The problem is that unless enough people upload their results to Phylotree, that subgroup will never be identified, so a new haplogroup won’t be added.
If the entire Family Tree DNA data base were to be uploaded to Phylotree, can you imagine how many new haplogroups would need to be formed? Of course, Family Tree DNA can’t do that, but individual testers can and should.
Challenges for Vendors
The challenge for vendors is that every time the phylotree tree is updated and a new version is produced, the vendors must “rerun” their existing tester samples against the new haplogroup defining mutations to update their testers’ haplogroup results.
In some cases, entire haplogroups are obsoleted and branches moved, so it’s not a simple matter of just adding a single letter or digit. Rearranging occurs, and will occur more and more, the more tests that are uploaded to Phylotree.
For example, in the Phylotree V17 update, haplogroup A4a1 became A1a. In other words, some haplogroups became entirely obsolete and were inserted onto other branches of the tree.
In the current version of the Phylotree, haplogroup A4 has been retired.
Keep in mind that all haplogroup assignments are the cumulative combination of all of the upstream direct haplogroups. That means that haplogroup A4a1, in the prior version, had all of the haplogroup defining mutations shown in bold in the chart below. In the V17 version, haplogroup A1a contains all of the mutations shown in bold red. You might notice that the haplogroup A4 defining mutation T16362C is no longer included, and haplogroup A4, plus all 9 downstream haplogroups which were previously dependent on T16362C have been retired. A4a1 is now A1a.

Taking a look at the mitochondrial tree in pedigree fashion, we can see haplogroup A4a1 in Build 15 from September 2012, below.

Followed by haplogroup A1a in the current Build 17.

Full Sequence Versus Chip Based Mitochondrial Testing
While Family Tree DNA tests the full sequence of their customers who purchase that level of testing, other vendors don’t, and these changes wreak havoc for those vendors, and for compatibility for customer attempting to compare between data bases and information from different vendors.
That means that without knowing which version of Phylotree a vendor currently uses, you may not be able to compare meaningfully with another user, depending on changes that occurred that haplogroup between versions. You also need to know which vendor each person utilized for testing and if that vendor’s mitochondrial results are generated from an autosomal style chip or are actually a full mitochondrial sequence test. Utilizing the ISOGG mtDNA testing comparison chart, here’s a cheat sheet.
| Vendor | No Mitochondrial | Chip based haplogroup only mitochondrial | Full Sequence mitochondrial |
| Family Tree DNA | No | Yes – V17 | |
| 23andMe | Yes – Build V7 | No | |
| Ancestry | None | ||
| LivingDNA | Yes – Build V17 | No | |
| MyHeritage | None | ||
| Genographic V2 | Yes – Build V16 | No |
Of the chip-based vendors, 23andMe is the most out of date, with V7 extending back to November of 2009. The Genographic Project has done the best job of updating from previous versions. LivingDNA entered the marketplace in 2016, utilizing V17 when they began.
Family Tree DNA’s mitochondrial test is not autosomal chip based, so they don’t encounter the problem of not having tested needed locations because they test all locations. They have upgraded their customers several times over the years, with the current version being V17.
Family Tree DNA’s mitochondrial DNA test is a separate test from their Family Finder autosomal test while the chip-based vendors provide a base-level haplogroup designation that is included in their autosomal product. However, for chip-based vendors, updating that information can be very challenging, especially when significant branch changes occur.
Let’s take a closer look.
Challenges for Autosomal Chip-Based Vendors Providing Mitochondrial Results
SNP based mitochondrial and Y DNA testing for basic haplogroups that some vendors include with autosomal DNA is a mixed blessing. The up side, you receive a basic haplogroup. The down aide, the vendor doesn’t test anyplace near all of the 16,569 mitochondrial DNA SNP locations.
I wrote in detail about how this works in the article, Haplogroup Comparisons Between Family Tree DNA and 23andMe. Since that time, LivingDNA has also added some level of haplogroup reporting through autosomal testing.
How does this work?
Let’s say that a vendor tests approximately 4000 mitochondrial DNA SNPs on the autosomal chip that you submit for autosomal DNA testing. First, that’s 4000 locations they can’t use for autosomal SNPs, because a DNA chip has a finite number of locations that can be utilized.
Secondly, and more importantly, it’s devilishly difficult to “predict” haplogroups at a detailed level correctly. Therefore, some customers receive a partial haplogroup, such as J1c, and some receive more detail.
It’s even more difficult, sometimes impossible, to update haplogroups when new Phylotree versions are released.
Why is Haplogroup Prediction and Updating so Difficult?
The full mitochondrial DNA sequence is 16,569 locations in length, plus or minus insertions and deletions. The full sequence test does exactly what that name implies, tests every single location.
Now, let’s say, by way of example, that location 10,000 isn’t used to determine any haplogroup today, so the chip-based vendors don’t test it. They only have room for 4000 of those locations on their chip, so they must use them wisely. They aren’t about to waste one of those 4000 spaces on a location that isn’t utilized in haplogroup determination.
Let’s say in the next release, V2, that location 10,000 is now used for just one haplogroup definition, but the haplogroup assignment still works without it. In other words, previously to define that haplogroup, location 9000 was used, and now a specific value at location 10,000 has been added. Assuming you have the correct value at 9,000, you’re still golden, even if the vendor doesn’t test location 10,000. No problem.
However, in V3, now there are new haplogroup subgroups in two different branches that use location 10,000 as a terminal SNP. A terminal SNP is the last SNP in line that define your results most granularly. In haplogroup J1c2f, the SNP(s) that define the f are my terminal SNPs. But if the vendor doesn’t test location 10,000, then the mutation there can’t be used to determine my terminal SNP, and my full haplogroup will be incomplete. What now?
If location 10,000 isn’t tested, the vendor can’t assign those new haplogroups, and if any other haplogroup branch is dependent on this SNP location, they can’t be assigned correctly either. Changes between releases are cumulative, so the more new releases, the further behind the haplogroup designations become.
Multiple problems exist:
- Even if those vendors were to recalculate their customer’s results to update haplogroups, they can’t report on locations they never tested, so their haplogroup assignments become increasingly outdated.
- To update your haplogroup when new locations need to be tested, the vendor would have to actually rerun your actual DNA test itself, NOT just update your results in the data base. They can’t update results for locations they didn’t test.
- Without running the full mitochondrial sequence, the haplogroup can never be more current than the locations on the vendor’s chip at the time the actual DNA test is run.
- No vendor runs a full sequence test on an autosomal chip. A full mitochondrial sequence test at Family Tree DNA is required for that.
- Furthermore, results matching can’t be performed without the type of test performed at Family Tree DNA, because people carry mutations other than haplogroup defining mutations. Haplogroup only information is entertaining and can sometimes provide you with base information about the origins of your ancestor (Native, African, European, Asian,) but quickly loses its appeal because it’s not specific, can’t be used for matching and can’t reliably be upgraded.
The lack of complete testing also means that while Family Tree DNA can publish this type of tree and contribute to science, the other vendors can’t.
Let’s take a look at Family Tree DNA’s new tree.
Finding the Tree
To view the tree, click here, but do NOT sign in to your account. Simply scroll to the bottom of the page where you will see the options for both the Y DNA Haplotree and the mtDNA Haplotree under the Community heading.
Click on mtDNA Haplotree.
If you are a Family Tree DNA customer, you can view both the Y and mitochondrial trees from your personal page as well. You don’t have to have taken either the Y or mitochondrial DNA tests to view the trees.

Browsing the mtDNA Tree
Across the top, you’ll see the major haplogroups.
I’m using haplogroup M as an example, because it’s far up the tree and has lots of subgroups. Only full sequence results are shown on the tree.

The basic functionality of the new mitochondrial tree, meaning how it works, is the same as the Y tree, which I wrote about in the Family Tree DNA’s PUBLIC Y DNA Haplotree.
You can view the tree in two formats, countries or variants, in the upper left-hand corner. View is not the same thing as search.
When viewing the mitochondrial DNA phylotree by country, we see that haplogroup M has a total of 1339 entries, which means M and everything below M on the tree.
However, the flags showing in the M row are only for people whose full mitochondrial sequence puts them into M directly, with no subgroup.
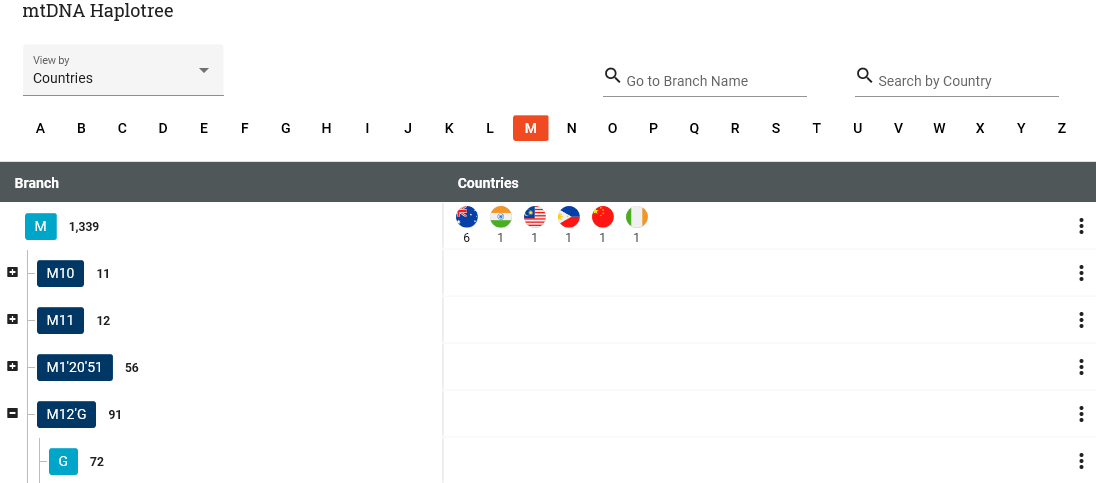
As you can see, there are only 12: 6 people in Australia, and one in 5 other countries. These are the locations of the most distant known ancestor of those testers. If they have not completed the maternal Country of Origin on the Earliest Known Ancestor tab, nothing shows for the location.
Viewing the tree by variant shows the haplogroup defining mutations, but NOT any individual mutations beyond those that are haplogroup defining.

For each haplogroup, click on the three dots to the right to display the country report for that haplogroup.

The Country Report
The Country Report provides three columns.
The column titled Branch Participants M shows only the total of people in haplogroup M itself, with no upstream or downstream results, meaning excluding M1, M2, etc. Just the individuals in M itself. Be sure to note that there may be multiple pages to click through, at bottom right.

The second column, Downstream Participants – M and Downstream (Excluding other Letters) means the people in haplogroup M and M subclades. You may wonder why this column is included, but realize that branches of haplogroup M include haplogroups G, Q, C, Z, D and E. The middle column only includes M and subgroups that begin with M, without the others, meaning M, M10, M11 but not G, Q, etc.
Of course the final column, All Downstream Participants – M and Downstream (Including other Letters) shows all of the haplogroup M participants, meaning M and all subclades, including all other haplogroups beneath M, such as M10, G, Q, etc..
What Can I Do with This Information?
Unlike the companion Y tree DNA, since surnames change every generation for maternal lineages, there is no requirement to have multiple matching surnames on a branch to be displayed.
Therefore, every person who includes a location for a most distant known ancestor is included in the tree, but surnames are not.
I want to see, at a glance, where the other people in my haplogroup, and the haplogroups that are the “direct ancestral line” of mine are found today. Clusters may mean something genealogically or are at least historically important – and I’ll never be able to view that information any other way. In fact, before this tree was published, I wasn’t able to see this at all. Way to go Family Tree DNA!!
It’s very unlikely that I’ll match every person in my haplogroup – but the history of that haplogroup and all of the participants in that haplogroup are important to that historical lineage of my family. At one time, these people all shared one ancestor and determining when and where that person lived is relevant to my family story.
Searching for Your Haplogroup
I’m searching for haplogroup J1c2f by entering J1c2f in the “Go to Branch Name.”

There it is.

I can see that there are 17 people in Sweden, 13 in Norway, 5 in Germany, 3 in Russia, etc. What’s with the Scandinavian cluster? My most distant known ancestor was found in Germany. There’s something to be learned here that existing records can’t tell me!
The mother branch is J1c2 which shows the majority of individuals in Ireland followed by England. This probably suggests that while J1c2f may have been born in Scandinavia, J1c2 probably was not. According to the supplement to Dr. Doron Behar’s paper, A “Copernican” Reassessment of the Human Mitochondrial DNA tree from its Root, which provides ages for some mitochondrial DNA haplogroups:
| Haplogroup | How Old | Standard Deviation | Approximate Age Range in Years |
| J1c2 | 9762 | 2010 | 7,752 – 11,772 |
| J1c2f | 1926 | 3128 | 500 – 5,054 |
I happen to know from communicating with my matches that the haplogroup J1c2f was born more than 500 years ago because my Scandinavian mito-cousins know where their J1c2f cousin was then, and so do I. Mine was in Germany, so we know our common ancestor existed sometime before that 500 year window, and based on our mutations and the mutation tree we created, probably substantially before that 500 year threshold.
Given that J1c2, which doesn’t appear to have been born in Scandinavia is at least 7,700 years old, we can pretty safely conclude that my ancestor wasn’t in Scandinavia roughly 9,000 years ago, but was perhaps 2,000 years, ago when J1c2f was born. What types of population migration and movement happened between 2,000 and 9,000 years ago which would have potentially been responsible for the migration of a people from someplace in Europe into Scandinavia.
The first hint might be that in the Nordic Bronze Age, trade with European cultures became evident, which of course means that traders themselves were present. Scandinavian petroglyphs dating from that era depict ships and art works from as far away as Greece and Egypt have been found.

The climate in Scandinavia was warm during this period, but later deteriorated, pushing the Germanic tribes southward into continental Europe about 3000 years ago. Scandinavian influence was found in eastern Europe, and numerous Germanic tribes claimed Scandinavian origins 2000 years ago, including the Bergundians, Goths, Heruls and Lombards.
Hmmm, that might also explain how my mitochondrial DNA, in the form of my most distant known ancestor arrived in Germany, as well as the distribution into Poland.
Is this my family history? I don’t know for sure, but I do know that the clustering information on the new phylotree provides me with clustering data to direct my search for a historical connection.
What Can You Do?
- Take a full mitochondrial DNA test. Click here if you’d like to order a test or if you need to upgrade your current test.
- Enter your Earliest Known Ancestor on the Genealogy tab of your Account Information, accessed by clicking the “Manage Personal Information” beneath your profile photo on your personal page.

The next few steps aren’t related to actually having your results displayed on the phylotree, but they are important to taking full advantage of the power of testing.
- While viewing your account information, click on the Privacy and Sharing tab, and select to participate in matching, under Matching Preferences.

- Also consent to Group Project Sharing AND allow your group project administrators to view your full sequence matches so that they can group you properly in any projects that you join. You full sequence mutations will never be shown publicly, only to administrators.

Of course, always click on save when you’re finished.
- Enter your most distant ancestor information on your Matches Map page by clicking on the “Update Ancestor’s Location” beneath the map.

- Join a project relevant to your haplogroup, such as the J project for haplogroup J. To join a project, click on myProjects at the top of the page, then on Join Projects.

- To view available haplogroup projects, scroll down to the bottom of the screen that shows you available projects to join, and click on the letter of your haplogroup in the MTDNA Haplogroup Projects section.
![]()
- Locate the applicable haplogroup, then click through to join the project.

These steps assure that you’ve maximized the benefits of your mitochondrial results for your own research and to your matches as well. Collaborative effort in completing geographic and known ancestor information means that we can all make discoveries.
The article, Working with Mitochondrial DNA Results steps you through you all of the various tools provided to Family Tree DNA testers.
Now, go and see who you match, where your closest matches cluster, and on the new mtDNA Haplotree, what kind of historical ancestral history your locations may reveal. What’s waiting for you?
______________________________________________________________
Disclosure
I receive a small contribution when you click on some of the links to vendors in my articles. This does NOT increase the price you pay but helps me to keep the lights on and this informational blog free for everyone. Please click on the links in the articles or to the vendors below if you are purchasing products or DNA testing.
Thank you so much.
DNA Purchases and Free Transfers
- Family Tree DNA
- MyHeritage DNA only
- MyHeritage DNA plus Health
- MyHeritage FREE DNA file upload
- AncestryDNA
- 23andMe Ancestry
- 23andMe Ancestry Plus Health
- LivingDNA
Genealogy Services
Genealogy Research
- Legacy Tree Genealogists for genealogy research
Share this:

Discover more from DNAeXplained - Genetic Genealogy
Subscribe to get the latest posts sent to your email.
L0A1B1A! I would surely love some matches at the full mitochondrial sequence level!!
that’s my dads oops mine is L0a1b1!
Did FTDNA get informed consent to use their customers’ data in a research study?
What study?
They have not done one themselves.
They haven’t moved my results over to Phylotree.
They have an undertaking to me as a customer to provide a haplogroup and they have done that privately and my details are not on view here.
My customer viewpoint is that with this publicly available tree they are just organising things anonymously and encouraging more people to test, which can only be good for using my results to find more about my all-female line, which is important to me.
I have elsewhere provided informed consent information to people for them to participate in medical trials, and I don’t see how that is relevant here.
Please explain.
Thanks so much for publishing Family Tree’s mtDNA haplotree. Appreciate it! Naturally, I went straight to my mtDNA haplogroup to look up the countries. I get excited when Family Tree notifies me of a new mtDNA match. Unfortunately, many people neglect to enter the country of origin of their most distant maternal ancestor. Thanks for explaining how to do that update. By the way, my mtDNA haplogroup is U6a7a1b.
My first cousin’s mtDNA haplogroup is j1c as tested by the National Geno project. (I’m related to him through his father, not his mom.) His mother’s ancestors were Lithuanian Jews. Do you know whether Jews can belong to the j1c2f mtDNA haplogroup? Lithuania is right across the Baltic from Scandinavia, Poland, and Germany.
I’ve not seen any Jewish results in J1c2f.
Perhaps he belongs to one of the other j1c branches.
Most people that subscribe to any of the GG sites do not enter their ancestral surnames, nor any ancestral locations, nor any tree, nor anthing at all. On most sites, particularly Ancestry, they use code names to remain essentially anonymous. Nothing ventured, nothing gained. Those that do so reveal nothing of themselves, yet they expect to be given something for nothing. However this is an endemic problem on all the sites and is best left to another thread of discussion.
As you may have noticed, most haplogroups classifications are exclusively male or female.
As for the Jewish question you posed about ethnicity and ‘belonging”, let’s address it by citing Groucho Marx: “Does that mean that Jews are allowed to go in only up to their knees?” (If anyone doesn’t know the quote I’m paraphrasing or understand the reference, just Google Groucho to find it.)
Thanks, Roberta. I sorta wish you had put the link to the tree earlier in the post – I never found it while I was looking on my own . . . or maybe this is your way to force the reader to read your entire article . . . 😉
I am a bit uncomfortable, however. Are the countries shown at each haplotype based on the “declared” earliest ancestor? If so, isn’t that highly misleading since it is based solely on the ability of the individual to accurately research their matrilinear line? The country report even has the footnote: “* All origins are self-reported by the participants and may not reflect accurate haplogroup origins.” It seems to me that this is another example of casual use of data that will lead people astray.
What else do they have to be able to use?
Oh, I totally agree – they have nothing else (that I am aware of). But does that make it right? If I only have a dollar in my pocket, should the store sell me the $100 shirt because “it’s all I have?” What’s the substantive difference between this presentation and the “haplomap” they use to show where their clientele are located?
Clusters are really important. Singletons not so much. Also, my most distant ancestor is from Germany. I live in the US. Big difference:)
Haplomap is for customers’ locations. Good for very recent connections, but useless for deep history.
These locations are for oldest known ancestors. Quite valuable for far back.
Generally I have found people’s own geographical attributions quite accurate here.
You may be showing signs of disappointment from another DNA lab’s recent release of autosomal information where that was derived from user-provided data.
Very disappointing for me too.
This is different.
The one or two instances still around of the oldest known relative in an all-female line having an obviously male name not-withstanding.
Yes, I am aware that the map is for the customers’ locations. And I recognize this presentation is for “earliest known ancestors’ locations.” my point was only that the reliability depends on the accuracy of the customer’s research and, even then, frequently does not go beyond a couple generations. Please note: I am one of the lucky ones here – my earliest known matrilinear ancestor was well documented in her early 17th century emigration from England to Massachusetts and her descendants similarly well documented on my particular branch. Several zero GD and 1GD matches tracing their own descent from different daughters of the same woman effectively confirmed each of our matrilinear trees. But many other 0GD matches have not, or cannot, get back there even though it is highly likely that they descend from the same woman. They name their “origin” as the US . . . of little use to anyone (although my info might be helpful to them). As always, your mileage may vary . . .
Thanks to our family genealogist, I was able to trace my matrilineal ancestor to my great-Great-great grandmother in the Ukraine. For other branches of my family, I can’t go beyond my grandparents!
Helix does full Sequence MtDNA and partners with Geno 2.0.
Sincerely, Marion.
Note:
The information in this electronic mail message is sender’s personal and/or business Confidential and may be legally privileged. Do not forward this email or discuss it with anyone without the sender’s permission. If you are not the intended recipient, any disclosure, copying, distribution or any action taken or omitted to be taken in reliance on it is prohibited and may be unlawful. By reading the message and opening any attachments, the recipient accepts full responsibility for taking protective and remedial action about viruses and other defects. The sender is not liable for any loss or damage arising in any way from this message or its attachments.
________________________________
Do you receive your full sequence haplogroup with your Nat Geo year? I didn’t think so.
I think I’m missing something critical. My FTDNA haplogroup is listed as H1b2a but when I browse H I do not see this group. In fact I do not see any groups with a number following the b. All groups show as two letters at this point for example: H1ba, H1bc, H1bd. What am I not understanding?
Enter H1b2a in “go to branch name.” It’s there. I just looked. The plus sign in front of the haplogroup then shows you the subgroups, or subgroup in this case.
Theresa, you are my father’s FTDNA mtDNA match, GD 3. My father’s maternal line is Finnish, known to the early 1600’s.
This was interesting. In my case my earliest maternal ancestor is my grandmother and it is a guess based on naturalization and census data. She died before I was born. In my haplogroup U8a1a1b1. There are 2 for poland . The highest two are 11 Sweden and 9 Finland. Would that mean possibly g grandmother and earlier were born in Sweden or Finland?
That’s a good possibility.
Roberta, Perfect timing. I am preparing a talk on mtDNA for next Tuesday for our local DNA Interest Group. I wanted to cite this and one other post and possibly snip parts of it for a powerpoint I am preparing. Of course, credit would be given to you. Do I have your permission?
Yes, and give them the blog link in case they’d like to subscribe:)
Absolutely.
Roberta,
On your October 4th post, please be aware that mitosearch was taken down on May 18th due to the new European privacy laws. I snipped the image but can’t post it here.
I wrote about that here: https://dna-explained.com/2018/05/14/world-families-network-ysearch-and-mitosearch-bite-the-dust-thanks-so-much-gdpr/
Thanks Roberta. This is helpful. My mother line is the extremely rare Haplogroup “X” (X2d1a) from northern Italy. So far, there are only 6 matches: 2 from Italy, 2 from Ireland, and 2 with unknown origin. That’s a small group of matches from all that have tested. What might help put things into perspective is if FT DNA could post some general data of a) how many total my tests, b) how many full sequence Mt tests, and c) how many full sequence without point of origin.
I too get frustrated with folks not posting their most distant known ancestors, even though I do realize some folks feel uncomfortable with that information being made available to matches. But what makes me more frustrated is the sheer number of hv1 and hv2 matches who haven’t upgraded to full sequence since hv1 and hv2 aren’t very useful for genealogy unless the tester has posted their most distant known ancestors (a vicious cycle!)
Anyway, I think it is a great start and I hope someday FT DNA will make the my Haplotree available on the tester’s results page, like the Y-haplotree is.
Might I posit that the “sheer number of hv1 and hv2 matches who haven’t upgraded to full sequence” is a function of the presentation by FTDNA to its potential customer base. I’d suggest that the vast number of those considering mtDNA testing are put off by the cost relative to the potential benefit, and that FTDNA is aware of that reaction. Accordingly they offer a couple lower priced products and many purchase those because “they want to test their maternal line” without realizing that the usefulness is effectively non-existent. MtDNA testing is clearly the stepchild in the testing world, coming way after autosomal testing and substantially after Y-dna testing in it’s appeal for both reward and cost. If FTDNA were to drop the two low-end tests and then lower the full sequence price, I suspect there would be a jump in participants.
The history is that for many years, the HVR1 and 2 were the only tests available. Lots of those early testers haven’t upgraded.
In some cases, because they are now with their ancestors, unfortunately.
All the GG sites depend upon profitability to continue to be ‘a going concern’. For those who might not understand that prase it means that a constant flow of income from new customers is needed for them to remain in business.
@ Mr. Fulllerton: I agree that your suggestions would benefit those of us who might benefit from your proposals. But the GG companies have their principal interests to pursue, and the big three dominate the GG field. In other words, I agree that it would be nice if they followed your suggestions for FTDNA, but it is not likely to happen. Ultimately the major players exist by making a profit, rather than putting the needs of genealogists first. Thus, the serious genealogist will always be a “stepchild in the testing world”. IMO, we need to accept that there are currently three major competitors in the GG field, and one also dominates the field of traditional genealogy in providing access to records by fee subscription. All these sites are funded by those who are willing to pay for their services, and most who are in their paying customer bases are not genealogists, and usually not even amateur ones. If the day ever comes when there is no competition among these major sites, (were some or all to go out of business, and then a monopoly might occur) there is no guarantee that the service provided would be any better, nor any more easily accessible than that which exists now.
Yes, I also thought that a price adjustment,
some directed educational mailings, combined with a directed upgrade promotion would help convince more HV1 & HV2 members to upgrade to FMS.
I sure agree with that.
I’m really glad I took the FMS mtDNA test. It was worth the extra money because I found out that I’m descended from Sephardic Jews from Spain, which is a long ways from Ukraine, where my maternal ancestors were from.
Hello Roberta
Thank you very much for your explanation of the new FTDNA
– mitochondrial DNA pages that can be found at:
https://www.familytreedna.com/public/mt-dna-haplotree/L
Today the ‘RSRS Country report’ gives the total of FGS tests
performed by FTDNA is an impressive 153,128.
Thank you also for including the link to the page on my website
http://www.ianlogan.co.uk/submission.htm
which details how a FTDNA customer may upload their FGS – FASTA file
to the GenBank database.
The phylotree at http://www.phylotree.org is based largely on the data
held in the GenBank database; which today has about 45,504 sequences.
This simple total can be found using:
(includes about 1,000 duplicates, and some ancient sequences):
https://www.ncbi.nlm.nih.gov/nuccore?term=%22homo+sapiens%22+%5Borganism%5D+%22complete+genome%22+mitochondrion
Of the 45,504 sequences, just over 4,000 were uploaded by FTDNA in 2012,
and over 2,000 have been submitted by individual FTDNA customers,
one by one, following the details given on my webpage.
The new FTDNA-mtDNA database and the GenBank database do complement each other;
with the GenBank database having many more Asian and African sequences
than are found in the FTDNA-mtDNA database.
Looking at all mitochondrial DNA Haplogroups is very interesting,
however, can I just mention 3 instances which are particularly so:
King Richard III’s family is in Haplogroup J1c2c3;
and the new FTDNA-mtDNA database shows that only 13 sequences
have been found so far. All of which are presumed to go back to England
in the early Middle Ages.
Haplogroup U6a7a1a is Acadian (French-Canadian) and have a defining
mutation that might be beneficial to members; but just how is uncertain.
The new FTDNA-mtDNA database shows that 72 people have been tested.
‘Polycythaemia Vera’, an uncommon blood disorder, may well have
a maternal line origin; and only be found in people from certain
mitochondrial Haplogroups. But which ones ?
To learn more, perhaps look at: http://www.ianlogan.co.uk/pv-et/start.htm
Ian Logan
So good o hear from you Ian. I’ll be writing shortly about how to upload one’s full sequence data.
I will be looking forward to this post. I gave my talk on mtDNA last night and referenced your blog several times and in the acknowledgements. I’m going to email them three links from dna-explained. You make a great contribution to all of us.
dang so many countries listed for b2
Thanks so much, Roberta, for posting this. Sent my mtdna data tonight—hopefully correctly! BTW, I’m J1c2. Hoping to find my long-long maternal line cousins. Thanks, again, for the great job you do here. Always enjoy reading your excellent blog!
Waiting for results is the hardest part.
Pingback: Whole Genome Sequencing – Is It Ready for Prime Time? | DNAeXplained – Genetic Genealogy
Pingback: Thirteen Good Reasons to Test Your Mitochondrial DNA | DNAeXplained – Genetic Genealogy
Pingback: Mitochondrial DNA: Part 2 – What Do Those Numbers Mean? | DNAeXplained – Genetic Genealogy
Pingback: Family Tree DNA Dashboard Gets a New Skin | DNAeXplained – Genetic Genealogy
Pingback: The Million Mito Project | DNAeXplained – Genetic Genealogy